Osmotic demyelination syndrome caused by rapid correction of hyperammonemia and continuous hyperbilirubinemia: a case report and review of the literature
Article information

Abstract
Osmotic demyelination syndrome (ODS) is an acute demyelinating disorder characterized by the loss of myelin in the center of the basis pons, defined as central pontine myelinolysis (CPM), and demyelination in locations outside the pons, defined as extrapontine myelinolysis (EPM). ODS including CPM and EPM is mainly caused by rapid correction of hyponatremia. However, there are several reports of ODS in medical conditions such as malnutrition; alcoholism; liver transplantation; malignancy; sepsis; and electrolyte imbalance including hypernatremia, hypokalemia, hypophosphatemia, and chronic illness. ODS caused by rapid correction of hyperammonemia or continuous hyperbilirubinemia without sodium fluctuations has rarely been reported. Because ODS may be irreversible, prevention is crucial. Herein, we report a case of ODS secondary to rapid correction of hyperammonemia and continuous hyperbilirubinemia.
Introduction
Osmotic demyelination syndrome (ODS) is an acute demyelinating disorder in the central nervous system and occurs in patients with chronic hyponatremia [1,2]. In several case reports, ODS was associated with liver transplantation, hypernatremia, hypokalemia, hypophosphatemia, hemodialysis, refeeding syndrome, malnutrition, diabetes mellitus, adrenal insufficiency, leukemia, lymphoma, systemic lupus erythematosus, acquired immunodeficiency syndrome, and sepsis [3-5]. ODS is commonly caused by rapid correction of hyponatremia over only a few hours and rarely occurs when hyperbilirubinemia is prolonged or hyperammonemia is rapidly corrected within normal serum sodium concentration range [6,7].
Herein, we report a case of ODS in a patient with rapid correction of hyperammonemia and continuous hyperbilirubinemia without changes in serum sodium level.
This study was approved by the Institutional Review Board of the Kyung Hee University Medical Center (No. 2009-12-301), and written informed consent was obtained for publication of this report and accompanying images.
Case Report
A 34-year-old male patient presented with loss of consciousness and was admitted to our hospital. His initial blood pressure was 124/65 mmHg. He had no family history or past history but consumed three bottles of alcohol per day. On initial neurological examination, his consciousness was stupor, and an avoidance response to pain stimulus was observed symmetrically. Brainstem reflexes were normal. Localizing or lateralizing neurologic deficit was not observed. The initial Glasgow Coma Scale score was 7 (E1V2M4).
The initial blood test results showed hyperammonemia (253.7 µmol/L) and hyperbilirubinemia (total bilirubin, 155.44 μmol/L; direct bilirubin, 81.23 μmol/L). The initial blood test results showed normal aspartate aminotransferase of 2,293 U/L, alanine aminotransferase of 304 U/L, alkaline phosphatase of 64 U/L, gamma-glutamyl transpeptidase of 276 U/L, protein of 4.8 g/dL, albumin of 1.9 g/dL, amylase of 730 U/L, lipase of 73 U/L, blood urea nitrogen of 15 mg/dL, creatinine of 1.04 mg/dL, and prothrombin time of 62.7 seconds. Electrolyte levels also were normal. The international normalized ratio was 3.18. A cerebrospinal fluid (CSF) test was performed to exclude encephalitis, and the results were the following: CSF pressure, 220 mmH2O; color, clear; red blood cells, 1/μL; white blood cells, 1/μL; protein, 53 mg/dL; and glucose, 88.8 mg/dL (serum glucose, 135 mg/dL). Specific findings associated with infections caused by coronavirus disease 2019, influenza virus, and other pathogens were not observed. In addition, immunoglobulin G index and aquaporin-4 antibody results were within the reference ranges. Abdominal computed tomography revealed the presence of moderate to severe ascites. Initial diffusion-weighted magnetic resonance imaging (MRI) of the brain was unremarkable (Figure 1). Due to the absence of lateralizing and localizing signs, metabolic encephalopathy resulting from liver failure was also considered.

Initial findings of diffusion-weighted magnetic resonance imaging
No abnormal lesion at (A) bilateral caudate nucleus, putamen, thalami, (B) midbrain, (C) central pons, or (D) bilateral middle cerebellar peduncle.
A lactulose enema was initially used to control hyperammonemia (correction rate, 83.6 μmol/L/day), and the level was normal (56.4 μmol/L) on the third day of hospitalization. However, the patient deteriorated to a semicomatose state and did not show vestibulo-ocular reflex response. Progressive spastic quadriparesis not initially observed during neurological examination was observed. Electroencephalography results showed continuous generalized slowing. On the 14th day, follow-up T2-weighted and diffusion-weighted MRI of the brain revealed elevated signal intensities in the bilateral caudate nucleus, putamen, thalami, midbrain, central pons, and bilateral middle cerebellar peduncle (Figure 2). The lesion without mass effect and contrast enhancement appeared hypointense on T1-weighted MRI. These findings were new compared with brain MRI results obtained on admission. During hospitalization, electrolyte levels including sodium remained in the normal range. However, hyperbilirubinemia persisted during the treatment period (total bilirubin range, 123.12–406.3 μmol/L). We suspected ODS was caused by rapid correction of hyperammonemia and persistent hyperbilirubinemia secondary to no change in serum sodium level. Consciousness remained semi-coma, and hyperbilirubinemia was observed during conservative management. His family did not agree to additional hemodialysis or liver transplant. Eventually, the patient died on the 76th day of hospitalization due to severe metabolic acidosis.
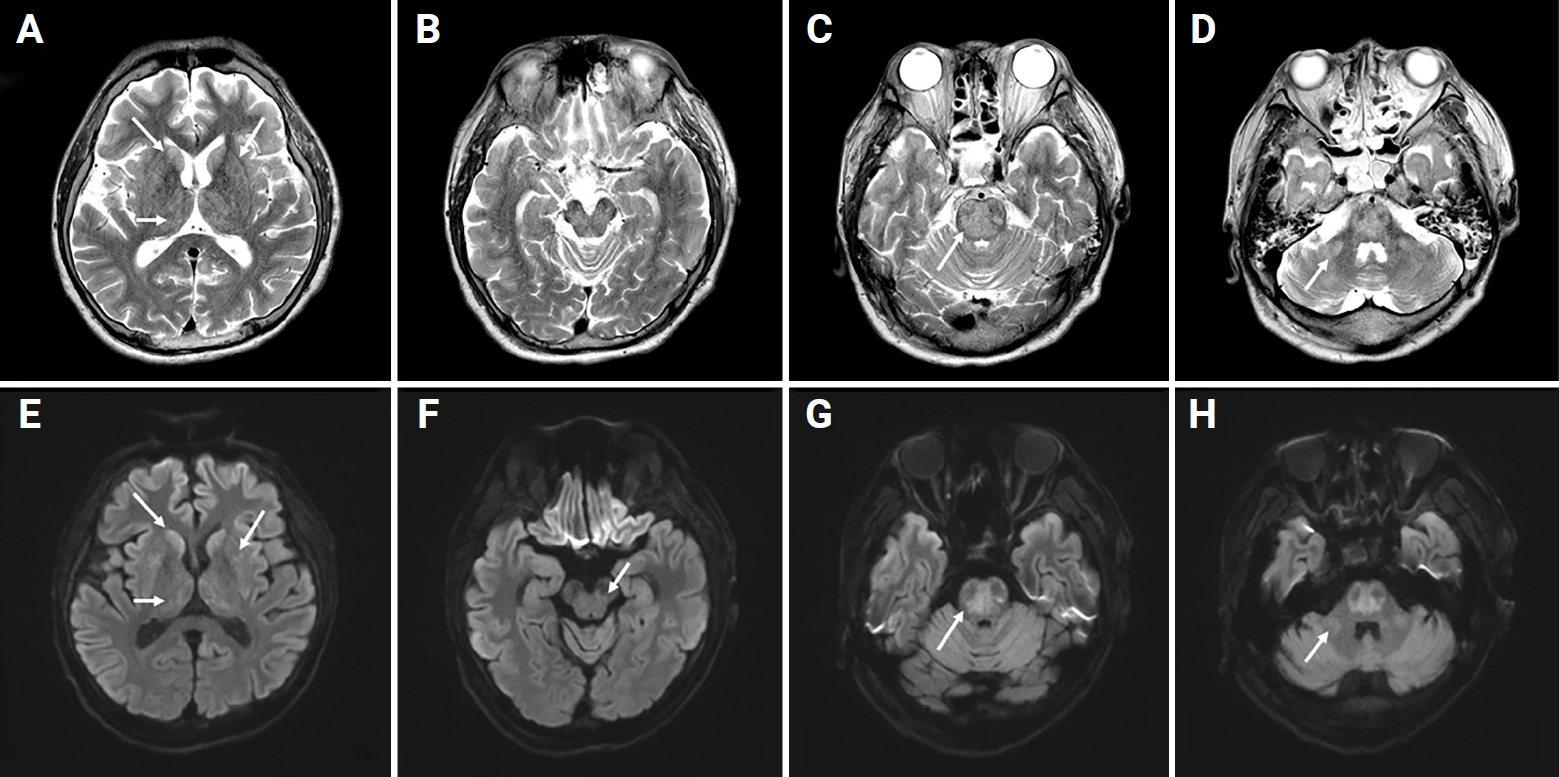
Brain T2-weighted and diffusion-weighted magnetic resonance imaging on the 14th day.
Osmotic demyelination syndrome (arrows) at (A and B) bilateral caudate nucleus, putamen, thalami, (C and D) midbrain, (E and F) central pons, and (G and H) bilateral middle cerebellar peduncle.
Discussion
The current case shows a patient with ODS that occurred at the bilateral caudate nucleus, putamen, thalami, midbrain, central pons, and bilateral middle cerebellar peduncle due to rapid correction of hyperammonemia and continuous hyperbilirubinemia without changes in serum sodium level.
ODS is rare and reported in 1% of patients admitted to a neurology department and 0.06% of all admissions to a general medical department. ODS is more common in adults than in children and has similar prevalence in males and females [8]. In general, ODS may present with a wide range of symptoms, including progressive spastic quadriparesis, pseudobulbar palsy, pseudobulbar affect, dysarthria, dysphagia, ophthalmoplegia, ataxia, nystagmus, and cranial nerve palsies [9]. Before presenting with ODS symptoms, patients may have neurological symptoms of encephalopathy. These symptoms usually resolve following initial correction of electrolytes.
ODS is commonly caused by rapid sodium correction of more than 12 mEq/L per day. Rapid correction of hyponatremia results in a rapid change in osmotic gradient, causing moisture in the cells to escape into the extracellular space and damage the vascular endothelium and glial cells that comprise the blood-brain barrier (BBB) [10]. When the BBB is damaged, inflammatory mediators such as cytokines, lymphocytes, complement, and vasoactive amines flow into the central nervous system, resulting in inflammatory demyelination (i.e., ODS) [4]. However, in the present case, the blood sodium level was within the normal range and did not fluctuate during the treatment period.
To estimate the rarity of this case, we reviewed all reported ODS cases from 1960 to 2023 based on a search of English language literature using PubMed and Google Scholar databases. The literature was searched using the following terms: osmotic demyelination syndrome, ODS, central pontine myelinolysis, CPM, extrapontine myelinolysis, EPM, correction of ammonia, hyperammonemia, and hyperbilirubinemia. Very few cases of ODS, CPM, or EPM were reported in patients with rapid correction of hyperammonemia or continuous hyperbilirubinemia and are summarized in Table 1. In children, most cases of hyperammonemia were caused by genetic abnormalities including ornithine transcarbamylase deficiency; however, most cases in adults were caused by chronic alcoholism [11-17]. MRI findings in most ODS reports were T2 hyperintensity in the central pons, with sparing of the ventrolateral pons, tegmentum, and corticospinal tracts. Contrast enhancement was usually not observed. Pontine lesions were usually symmetrical and extrapontine lesions bilateral and most commonly observed in cerebellar peduncles, globus pallidus, thalamus, lateral geniculate body, putamen, external capsule, splenium of the corpus callosum, and supratentorial white matter [18]. Rapid correction of hyperammonemia or continuous hyperbilirubinemia simultaneously caused both CPM and EPM [11-16]. Only one CPM case report due to continuous hyperbilirubinemia was identified [17].
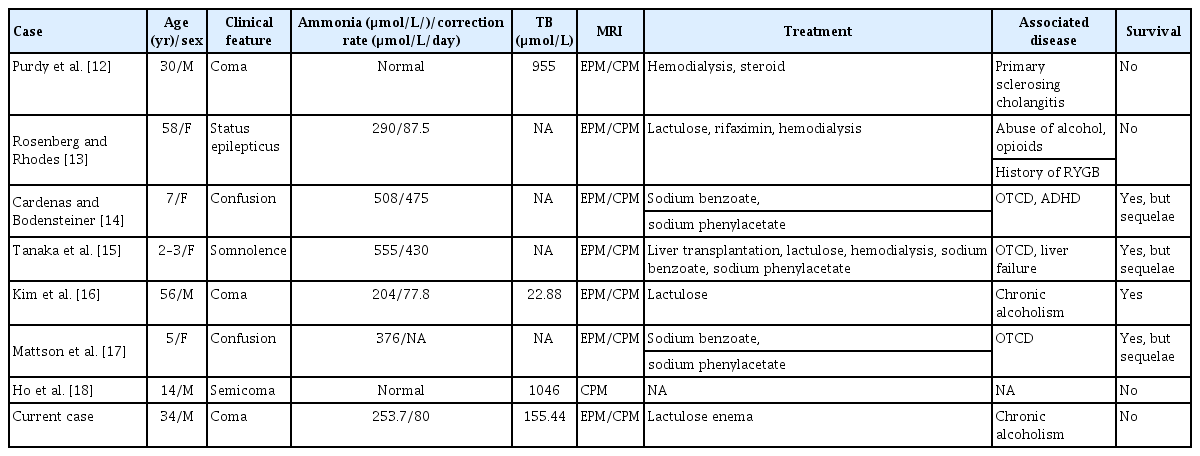
Reported cases of CPM and EPM caused by rapid correction of hyperammonemia or continuous hyperbilirubinemia
In the literature, a quick correction of ammonia lower than 77.8 μmol/L/day was associated with a nonlethal prognosis. In previous studies, the directly calculated changes in osmolarity due to changes in ammonia concentration over a 24-hour period were compared with the safe range of osmolarity change rates due to alterations in serum sodium concentration. The resulting osmolarity change rates directly caused by ammonia correction (previous cases: −0.459, −0.220, lower than −0.376 mOsm/L/day and our case: −0.077 mOsm/L/day) were negligible in absolute value compared with the safe range of osmolarity change rates due to sodium concentration [13,14,16]. Therefore, factors other than osmolarity change rate directly caused by ammonia were suggested as causes of ODS. Ammonia upregulates aquaporin-4 in astrocytes and penetrates the BBB, resulting in edema of astrocytes. Sudden correction of ammonia may lead to aquaporin-4 dysfunction similar to chronic neuroinflammatory diseases such as multiple sclerosis and neuromyelitis optica, leading to demyelinating lesions due to osmotic stress [1,15].
Furthermore, in the literature, ammonia levels higher than 204 μmol/L/day were associated with a lethal prognosis. In patients with hyperammonemia, glutamate plays a role in reducing intracellular ammonia by decreasing intracellular space and increasing extracellular space in the brain [19]. Glutamate is an important osmolyte that is mainly released from cells when cell volume needs to be reduced, contributing to the reduction of intracellular osmolarity [20]. In patients with hyperammonemia, the mechanism described above can reduce the difference in glutamate intracellular/extracellular concentration, which may contribute to the disturbance of cell volume regulatory mechanisms [13,14]. In addition, continuous hyperammonemia can cause numerous neurotoxic effects such as cerebral edema, brain herniation, and death. However, a significant correlation has not yet been established between CPM and hyperammonemia [21].
Continuous hyperbilirubinemia can also cause ODS. In the early stage of hyperbilirubinemia, secretion of interleukin or vascular endothelial growth factor from microvascular endothelial cells is suppressed [8,22]. However, when hyperbilirubinemia persists for an extended period of time, the production and secretion of cytokines and nitric oxide are promoted in vascular endothelial cells. Because the production of nitric oxide is increased in a time- and concentration-dependent manner, the elevated permeability increases with the length of duration and the severity of hyperbilirubinemia [23]. Consequently, continuous hyperbilirubinemia weakens the BBB by causing damage to vascular endothelial cells, leading to ODS including CPM and EPM [11].
ODS and CPM caused by rapid correction of hyperammonemia or continuous hyperbilirubinemia showed a better prognosis in younger patients [13,14,16] and a poorer prognosis in most adult patients. Severe hyponatremia (less than 115 mEq/L), associated hypokalemia, or low Glasgow Coma Scale score (less than 10) at presentation or ODS following liver transplant were reported to be poor prognostic factors in ODS [24]. However, in our case report and based on the literature review, ODS and CPM caused by rapid correction of hyperammonemia or continuous hyperbilirubinemia had a poor prognosis in adult patients.
In the present case, rapid correction of hyperammonemia without fluctuation of sodium level was performed on the third day of admission, and extensive lesions in the bilateral caudate nucleus, putamen, thalami, midbrain, central pons, and both middle cerebellar peduncles were newly observed on follow-up brain MRI on the 14th day. Although lesions affecting the basal ganglia, caudate nucleus, thalamus, cerebellum, and subcortical white matter are frequently observed in hyperammonemic encephalopathy; mainly insula, cingulate gyrus, and extensive cortical lesions are observed in hyperammonemic encephalopathy [25,26]. However, in the present case, insula, cingulate gyrus, and extensive cortical lesions were not observed on brain MRI. In addition, symmetrically increased signal intensity of the globus pallidus, subthalamic nuclei, and hippocampus was observed on T1-weighted images at the acute stage of bilirubin encephalopathy [27,28]. However, symmetrical high signal intensities in the globus pallidus, subthalamic nuclei, and hippocampus were not observed in our patient. Therefore, we concluded that the cause was rapid correction of hyperammonemia or continuous hyperbilirubinemia.
Because serum sodium level was continuously in the normal range and hyperammonemia was only found in the early stages of treatment, ODS caused by continuous hyperbilirubinemia could have been suspected after identifying progressive spastic quadriparesis and ophthalmoplegia. Continuous hyperbilirubinemia was observed during the treatment period (range of total bilirubin, 123.12–406.3 μmol/L), and a neurological deficit was persistent. In previous reports, high-dose methylprednisolone was initiated to improve the integrity of the BBB by blocking inflammatory mediators [29]. However, clinical improvement was not observed. Furthermore, plasma exchange was shown to improve clinical outcomes in patients with ODS caused by continuous hyperbilirubinemia [30]. However, the family of our patient did not want him to receive plasma exchange treatment.
Although the change in sodium level is usually not significant, ODS can occur in patients with liver failure due to rapid correction of hyperammonemia or continuous hyperbilirubinemia. Therefore, physicians should pay attention to preventing ODS when treating patients with liver failure. Because this was a single case report and literature review, cross-sectional retrospective observational studies are needed to gain more knowledge regarding ODS caused by rapid correction of hyperammonemia or continuous hyperbilirubinemia.
Notes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Author Contributions
Conceptualization, Data curation: all authors; Formal analysis, Investigation, Methodology, Resources, Software, Visualization: Park K, Woo HG; Project administration: Woo HG; Supervision: Kim SB, Yoon SS; Writing–original draft: Park K, Woo HG; Writing–review and editing: Kim SB, Yoon SS, Woo HG
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. RS-2023-00239251 to HGW).